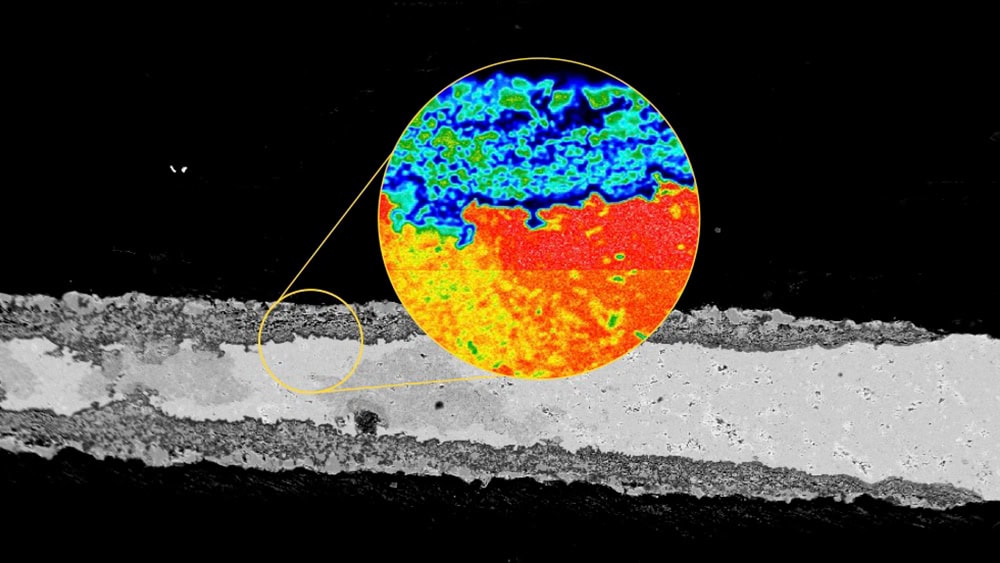
[Image above] Electron micrograph of a MAX phase material. MAX phases demonstrate high-temperature corrosion and oxidation resistance, which give them a lot of potential in several industries, including nuclear power and aerospace propulsion systems. Credit: Miladin Radovic, Texas A&M Engineering
On Tuesday, we highlighted recent work being done on MAX phases, a class of layered ceramic materials not often covered on CTT. These materials exhibit both high-temperature corrosion and oxidation resistance, which makes them ideal candidates for use in several industries, including nuclear power and aerospace propulsion systems.
MAX phase materials have the general formula Mn+1AXn, where M is a transition metal (toward the left-hand side of the periodic table), A is a metalloid (edging into the left-hand transition metals and heavy metals), and X is carbon or nitrogen. Researchers have studied more than 150 compositions with this general formula to date, with a big focus on ones containing titanium. (Studies on the Cr–Al–C system are slowly accumulating as well, as the Tuesday CTT showed.)
The number of known MAX phases continues to increase regularly thanks to a combination of experimental works and theoretical calculations. However, not all possible compositions are thermodynamically stable—at elevated temperatures and under oxidizing conditions, many MAX phases undergo detrimental self-sustaining oxidation reactions that result in catastrophic loss of mechanical integrity.
Because the oxidation process is complex, it is both difficult and expensive to model or characterize the process with current computational and experimental methods, thus slowing the process of identifying desirable MAX phases. Fortunately, a recent open-access article by researchers at Texas A&M University describes a new computational tool that can vastly speed up the process.
The researchers are led by materials science and engineering professors Raymundo Arróyave and Miladin Radovic. In a Texas A&M press release, Arróyave explains why density functional theory, an established physics-based mathematical model frequently used to explain materials phenomena, struggles to evaluate the properties of MAX phases at different temperatures.
“These calculations scale very poorly,” he says. “For perspective, if we want to use density functional theory to calculate the properties of a candidate material at the lowest temperature of zero kelvins, that is at the ground state, it might take about a day of computational time. But now, if you want to calculate the same properties at a finite temperature, say 1000 kelvins, it can take weeks.”
In addition, predicting the behavior of materials when exposed to oxygen at elevated temperatures further complicates the situation and can cause density functional calculations that account for oxidation to take months or even longer, Arróyave adds.
To speed up the time it takes to model oxidation of different MAX phases at high temperatures, some studies have demonstrated that augmenting density functional calculations with machine learning and experimentally acquired information can quickly and accurately improve predictions. So, the researchers of the new study decided to augment density functional calculations using an emerging machine learning method called sure independence screening and sparsifying operator (SISSO) in combination with a computational thermodynamics technique called grand-canonical linear programming (GCLP).
In an email, Arróyave says they chose SISSO as their machine learning model because of its ability to clearly demonstrate connections between inputs and outputs. “Typically, machine learning models are more like ‘black boxes,’ with very unintuitive connections between inputs and outputs,” he says. “On the other hand, SISSO allows you to create models that can be written as a function of intrinsic materials properties. The outcome of such a model is an equation that can be explicitly written and then shared to anyone.”
To test their approach, the researchers first extracted data from a couple of external databases and performed density functional calculations to determine a few fundamental properties of MAX phases at zero kelvins. Then, they gathered this information into an internal database and input the data into the machine-learning-based scheme to determine the most stable compounds for a given temperature and a certain MAX phase composition.

The high-throughput predictions given by the machine-learning-based model for several MAX phases—Ti2SiC, Ti2AlC, and Cr2AlC—compared well with values determined via oxidation experiments, thus establishing “the generality of the scheme … [and] strength of our framework.”
For the minor disagreements that did arise between theory and experiment, the researchers say it is likely due to not including all possible non-stoichiometric compounds, ternary-oxides/oxycarbides, and/or contributions from kinetics in the model. “However, keeping the complexity of the problem in mind, the proposed scheme provides good quantitative agreement with experiments,” they emphasize.
Arróyave says they are working with colleagues who have expertise in artificial intelligence and machine learning to develop more robust frameworks capable of building more complex models than SISSO, which they will use to design novel alloys. “We are also using the same approach that we used in this paper to predict how other materials oxidize when exposed to high temperatures and oxidizing environments,” he adds.
The open-access paper, published in npj Computational Materials, is “High-throughput reaction engineering to assess the oxidation stability of MAX phases” (DOI: 10.1038/s41524-020-00464-7).


