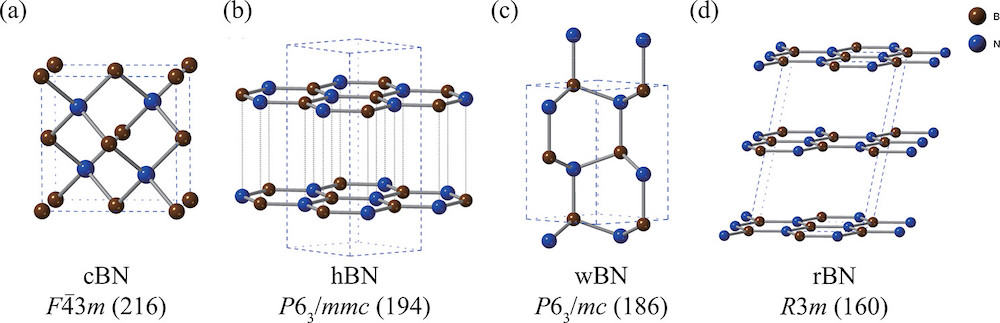
[Image above] Structures of (a) cubic zinc-blende, (b) hexagonal, (c) wurtzite, and (d) rhombohedral boron nitride. Hexagonal and rhombohedral are considered sp2-hybridized boron nitrides, while cubic zinc-blende and wurtzite are considered sp3-hybridized boron nitrides. Boron and nitrogen atoms are colored in brown and blue, respectively. Credit: Nikaido et al., The Journal of Physical Chemistry C (CC BY-NC-ND 4.0)
When designing new technologies, it’s not always about finding the right material for the job—sometimes it’s about finding the right material structure.
In materials science, polymorphism describes the existence of a solid material that can form in multiple different crystal structures. These different structures can lead a material to display very different properties even though the various forms (called polymorphs) are chemically identical.
Graphite and diamond are common examples of carbon polymorphs, with the former being very brittle and the latter being very hard. Zirconium carbide is another polymorph example of great interest to ceramic scientists, as discussed on CTT last month.
Boron nitride is a ceramic that forms in multiple stable structures, namely hexagonal (hBN), cubic zinc-blende (cBN), rhombohedral (rBN), and wurtzite (wBN). Of these, hexagonal boron nitride is generally regarded as the most stable form, and it has received much attention lately in the 2D materials field.
However, though hBN is generally regarded as the most stable structure, the relative phase stabilities of these polymorphs are still under debate.
“Solozhenko et al. claimed that cBN is more stable than hBN under ambient conditions, while Corrigan et al. and Fukunaga argued the opposite. Indeed, two conflicting results have been obtained, and it has been a long remaining question to clarify which of these experimental results is more reliable,” researchers write in a recent open-access paper.
The researchers are led by assistant professor Kousuke Nakano at the Japan Advanced Institute of Science and Technology and include colleagues from Oak Ridge National Laboratory, Indian Institute of Technology Madras, and S. N. Bose National Center for Basic Sciences.
They explain that many studies have used computational techniques to try and settle this debate. However, there are challenges in modeling these materials accurately.
“…the fact that hBN (and rBN) is a quasi-two-dimensional material makes it difficult to distinguish the relative stability of these polymorphs with standard mean-field ab initio calculations, because the van der Waals interaction cannot be evaluated quantitatively within LDA [local-density approximations] and other semilocal approximations that neglect the nonlocality of the exchange and correlation,” they write.
Functionals that include semiempirical van der Waals corrections can overcome this challenge within the density functional theory framework. However, results depend significantly on the type of correction used, as the researchers demonstrate once again through calculations in their paper.
Fortunately, new and refined computational methods that can aid in materials analysis are being continuously developed. One such method—quantum Monte Carlo simulations—has great potential for clarifying respective stabilities of boron nitride polymorphs.
Quantum Monte Carlo is a state-of-the-art ab initio framework that does not lose quantitative predictability, even for van der Waals solids. In particular, the fixed-node diffusion Monte Carlo (FNDMC) approach is effective at quantitatively treating van der Waals forces, and this method has been applied to many unusual compounds including graphite, black phosphorus, boron nitride bilayer, and molecular crystals.
Based on this potential, the researchers used FNDMC in their study to calculate the relative phase stabilities of the four boron nitride polymorphs. Their results support the general belief that hBN is the most stable structure. However, it also suggests that rBN is more stable than cBN, which a few other studies using sophisticated computational methods suggest as well.
The researchers note that while the overall FNDMC calculations they performed are robust, some uncontrollable errors do exist, such as fixed-node approximation and locality error.
“Investigating the effects of these uncontrollable errors on the binding energy calculations of the BN polymorphs is an interesting topic for future work, and it would provide a deeper insight into the relative stability of the BN polymorphs,” they write.
The open-access paper, published in The Journal of Physical Chemistry C, is “Diffusion Monte Carlo study on relative stabilities of boron nitride polymorphs” (DOI: 10.1021/acs.jpcc.1c10943).


